Warning of serious brain disorders in people with mild coronavirus symptoms.
Sofistek that is a scary notion, brain disease from mild disease. And I don’t question the findings as such. It may not be obvious to some (maybe to people here) but it is incredibly difficult to address issues in milder illness because these patients are sent home to recover once they test positive. We know a lot about ICU patients but our knowledge of ambulatory patients will take a long time to acquire. I believe the reason is the mandated isolated of patients, which is necessary, and shutdown of the outpatient centers where these workups are done. Mainly because of isolation requirements. This seems obvious but is having a big impact on what we know about mild disease. It could take years to get this data. Thank you for the article I know if I can find this information anywhere it will be here.
@nordicjack I do not believe in vaxxing. But for older people , and sick people , there may be no choice , even young people , there may be no choice. problem is older people do not build antobodies and react as strongly to vaccines ( flu ) as younger healthy people..There is always a choice on whether you're willing to inject aluminum into your own body. (Almost every single vaccine contains it, the manufacturers admit it, but criminally-negligent doctors don't tell you.) It's about the worst thing you could ingest. It's healthier to consume a SAD diet for a year, maybe 10 years, than to take a flu shot or other trojan-horsed adjuvanted vaccine.
@sofistek Warning of serious brain disorders in people with mild coronavirus symptoms.Where these people vaccinated (and thus their terrain/immunocompetence destroyed)? What's their vitamin D status? (Maybe even 'How long were they socially isolated?' because loneliness / psychological distress translates to chronically elevated baseline cortisol levels which translates to immune system shutting down.) Without those questions, never asked in clownworld, it's difficult to attribute causality to the virus itself.
It appears you conclude that SARS Cov-2 was engineered but not in manner described int the published studies that I cited.
Why do arguments against the theory of COVID-19 development in a laboratory never consider the pangolin virus GD Pangolin CoV?
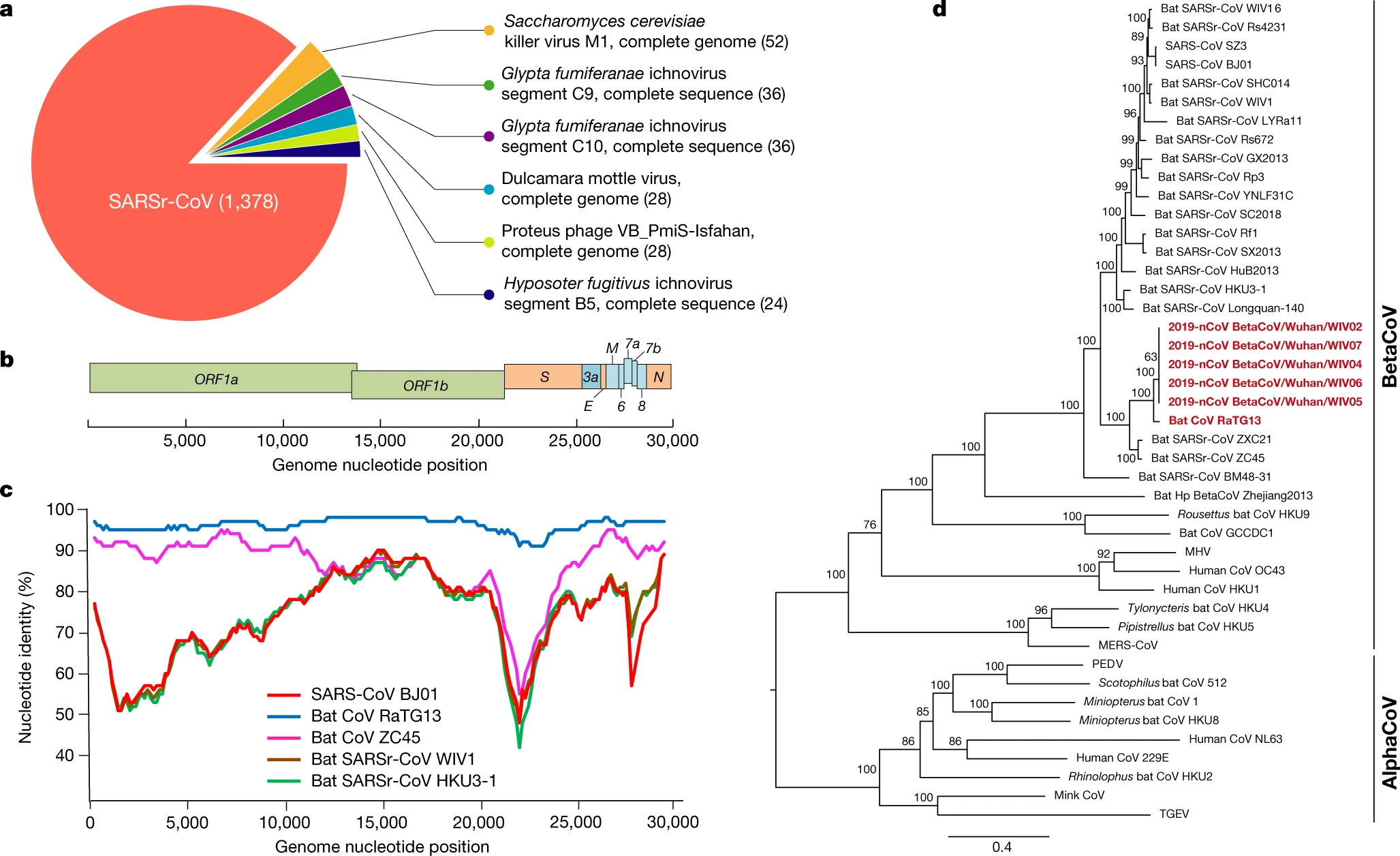
In Dr. Martenson’s discussion of the origin of the COVID-19 virus he compared the GD Pangolin CoV virus to COVID-19 (SARS-CoV-2)
“When we do, we discover that - hands down! - the all-important receptor binding domain (RBD) came from a pangolin, not a bat.
“It’s not even a contest. The pangolin/1 has 5 out of 5 all-important RBD contact amino acids (AAs - red boxes) in common with the pangolin, and 8 out of 9 adjacent AAs (yellow boxes). It’s closest ‘bat relative’ - RaTG13 only scores 1/5 for RBD contact, and 4/9 adjacent AAs. The other bat coronaviruses and SARS-classic are far worse.”
So why didn’t Daszak or Anderson et al. in their NatureMedicine article claim that COVID-19 developed naturally from the pangolin virus? They are silent on this possibility. Is it because pangolins don’t have communities like bats that permit passing viruses back and forth to develop mutations? According to Wikipedia, “Pangolins are solitary and meet only to mate.”
And they are endangered.
https://en.wikipedia.org/wiki/Pangolin
It is at a minimum equally likely to the “bat/pangolin” crossover hypothesis that the mutations from the pangolin virus was introduced into RATG13 by site directed mutagenesis on a plasmid encoding the entire viral genome. This a technique commonly used in molecular biology laboratories and is at a minimum equally likely. Also, consider how long both the pangolin virus and RATG13 was known by the chinese prior to SARS-COV-2…
Another way for RATG13 to accumulate the same mutations, would be by directed evolution. Here, the virus is cultured together with cells expressing human ACE2, only viruses with strong binding to human ACE2 will proliferate and rapidly mutations increasing binding to human ACE2 will accumulate.
I have been suspicious for a long while now and I support Chris on the lab origin. While it is rapidly dismissed by ordinary people as another conspiracy theory, I as a Molecular Biologist have had my suspicion for months… In my opinion we are looking at an accidental release, likely due to a lab technician or a scientist accidentally getting infected. They likely wrote it off as a common cold/flu and decided not to reveal their mistake… It is China after all and who knows what would happen to you if you make a stupid mistake. It wouldn’t be the first time a mistake happens in a lab and neither will it be the last…
Perhaps one of you could explain in terms that a non-molecular biologist can follow, I get the gist of what you are saying but I - and perhaps others in the community - would be interested in the family tree of SARS-COV2 and the derivation of its critical features.
Q: Why do arguments against the theory of COVID-19 development in a laboratory never consider the pangolin virus GD Pangolin CoV?
A: For clarification purposes, there is no such thing as the GD Pangolin CoV. GD is specifying Guangdong province of China. Malaysian Pangolins are typically smuggled into Guangdong, Guizhou, Guangxi, or Yunnan provinces but predominately into Guangdong to Shenzhen or Guangzhou cities. The relevant pCoV viruses that I believe you are referring to are actually from the Guangxi autonomous region. Those viruses are designated as GX. The GenBank accessions are PCoV_GX-P4L (MT040333), PCoV_GX-P1E (MT040334), PCoV_GX-P5L (MT040335), PCoV_GX-P5E (MT040336), PCoV_GX-P2V (MT072864). The Spike protein of RaTG13 has a higher match and furin score compared to any Pangolin virus on record, so we would deduce that the initial proximal zoonotic origin would be that of a bat and not a pangolin.
Q: In Dr. Martenson’s discussion of the origin of the COVID-19 virus he compared the GD Pangolin CoV virus to COVID-19 (SARS-CoV-2): “When we do, we discover that - hands down! - the all-important receptor binding domain (RBD) came from a pangolin, not a bat. It’s not even a contest. The pangolin/1 has 5 out of 5 all-important RBD contact amino acids (AAs - red boxes) in common with the pangolin, and 8 out of 9 adjacent AAs (yellow boxes). It’s closest ‘bat relative’ - RaTG13 only scores 1/5 for RBD contact, and 4/9 adjacent AAs. The other bat coronaviruses and SARS-classic are far worse.”
A: This information is correct. The amino acid residues in question are L455, F486, Q493, S494,N501, and Y505. These six key positions within the Receptor Binding Domain portion of the Spike protein have been suggested to increase virulence as well as promote a firmer reception. The Malaysian Pangolin virus in question is actually pCoV-MP789 (MT121216) which was sampled from a wild Pangolin in southern Yunnan province, quite close to the Malaysian border. Although these key amino acid residues play a role with virulency, there are only part of a much larger puzzle. The abscense of these 6 residues would simply weaken the virulency of nCoV-2019. Think of these as a specialized icing on the cake. The S1/S2 section is the true gem of this virus. When scoring the RBD region we actually find that RaTG13 is 86.19% similar to nCoV-2019 whereas pCoV-MP789 is 86.64%. Breaking down the specific differences within the RBD of each virus plays out the entire picture. More importantly, the Spike protein of RaTG13 scores 93.15% similarity to nCoV-2019 compared to only 84.52% from pCoV-MP789. The latter being the second higher scoring Spike comparison to this novel virus. We should consider the potency of the the RaTG13 spike protein as more significant than pCoV-MP789 while simultaneously accepting the 6 amino acid residues within the RBD of pCoV-MP789 provide higher virulency. Does this make sense? Both have their merits and nCoV-2019 appears to be a blend of the two.
Q: So why didn’t Daszak or Anderson et al. in their NatureMedicine article claim that COVID-19 developed naturally from the pangolin virus? They are silent on this possibility.
A: RaTG13 is by far the closest match, across the board. You need to take all proteins into consideration here. We cannot cheery pick. The RBD section of pCoV-MP789 is the only nod we can provide and even that is only a 0.45% differential. Both the Spike protein (93.15/84.52) and the ORF1ab (96.52/90.36) are clear indicators that a Rhinopholus Affinis bat is the probable zoonotic origin of nCoV-2019. In zoonotic viral recombinant process, we would expect the overwhelming majority of source RNA to be from the origin and only a fractional minority to be from the secondary source (Pangolin). A(primary)+B(secondary)=C. Does this make sense? Given the existence of RaTG13, it would not even make sense to suggest a Malaysian Pangolin as the single source of this virus.
Hope this was helpful information ![]()
I appreciate you ideas and thoughts. Sounds like you have lots of experience in this field.
Can you give us your best guess on where the virus came from.
Then, in a separate section, discuss the reasons for your conclusions. (This might help to separate out the technical details that most of us cannot understand or follow easily.)
Perhaps one of you could explain in terms that a non-molecular biologist can follow, I get the gist of what you are saying but I - and perhaps others in the community - would be interested in the family tree of SARS-COV2 and the derivation of its critical features.
In terms of the family tree, we would need to really dig down. We are way way down in sarbecovirus, which is a lineage of betacoronaviruses, which is a genus of coronaviruses.
Let’s say there are 4 different root families for coronaviruses; Alpha, Beta, Gamma, Delta. Let’s then say that the overwhelming majority of live coronaviruses in the world stem from Beta. Once we dig into Beta we have 4 lineages; let’s call these A, B, C, D. Two of the common colds, hCoV-OC43 and hCoV0HKU1 come from A. SARS comes from B. MERS comes from C. D is boring. Here is a tree outlining what I just described.
Virtually all bCoVs come from bats as they are natural reservoirs for viruses. Various viruses can live in bats for their entire lives with little to no impact to the host. This is also why Bats and not Pangolins would be the likely “source” as the viruses tend to have physical impacts on Pangolins and shorter their life span.
In this photo, you can see a partial phylogenic tree on the right side sorting various viruses based on complete genomes (nucleic acids). We use databases and software tools to compare genomes and these trees are comparative groupings. You can see BetaCoVs and Alphas listed there. The purpose of this specific chart was to underscore the similarity of nCoV-2019 to RaTG13, ZXC21, and ZC45. All of three of those listed viruses have controversial histories and genomes. You can spot SARS-CoV on the top.
When it lists SARSr, that means SARS related virus. Capitalized letters after the initial description denote a city name. Capitalized first letter and lower case second letter indicate the name of the specie they retrieved samples from
Bat-SARSr-Rs672: SARSr (SARS related, but was previously classified as SL or SARS-like). CoV (coronavirus). Rs (Rhinopolus sinicus, also known as the Chinese rufous horseshoe bat which is found in the Yunnan province of China, India, Vietnam, and Nepal). 672 is the sample number.
Another example: Bat-SARSr-CoV-ZXC21 and ZC45. I will do an entire post on these crazy viruses but the breakdown here is that ZC stands for Zhoushan City in Zhejiang, China. Whereas ZXC21 was discovered at a nearby mountain, so to avoid confusion they designated that site as ZXC as opposed to ZC.
Here is a Beta tree that includes the MP789 pCoV (listed as Pangolin-CoV 2020, even though it is from 2019). This tree scoring is the same as the previous, comparing NA of entire genomes. The “groupings” should be obvious to spot.
In terms of the key features of nCoV-2019 compared to SARSr CoVs that would require a much longer response. The KEY takeaway is that nCoV-2019 has a unique RNA insertion at a VERY crucial position that plays a vital role. That “type” of mutation is absent in ALL other betacovs (except one but lets ignore that lol).
{kind=link}
{kind=link}
{kind=link}
Thank you Dr. Mayer. You have my deepest appreciation for what you are sharing with us.
Q: Can you give us your best guess on where the virus came from.
A: The most probable origin is a chimeric virus created in a laboratory. Which lab or country would be complete speculation.
I focus on the science and not the news. The science is telling me that this virus was put together. The basis of the virus “appears” to be the RaTG13 virus. Why? Because that is the closest relative to nCoV-2019. This virus comes from a specific bat species in southern China and is not sold or found in wet markets. The second closest relative is a Malaysian Pangolin. Although this animal is sold/consumed in wet markets, last year scientists discovered a new betacoronavirus found in a specific Malaysian Pangolin. Neither of these two viruses could form nCoV-2019 but if we were to merge these two viruses then we would be around 98.9% identical. Does that make sense? The Bat virus has key features that the Pangolin virus does not have, while it is also true that the Pangolin virus has features that the Bat virus does not have. nCoV-2019 possesses all of these “features” since the original genome sequence. Aside from all of that, nCoV-2019 has this 1 specific SUPERMAN feature that no other betacoronavirus possesses; including the aforementioned Bat and Pangolin viruses.
How about this; I write an essay and then hand it to you for annotations and edits. I am the primary source (Bat) and you are the secondary source (Pangolin). In Microsoft Word we can track changes and can determine who wrote what and where the overlaps are. There is also some gibberish inserted into the document because you and I are sort of bumping heads as our papers are being mushed together. That gibberish is useless. But while reviewing the paper, we discover an entire paragraph written in Japanese. You did not write that. I did not write that. The gibberish mush could not have created it. Where did that paragraph come from? Hopefully that makes sense. This key SUPERMAN feature in nCoV-2019 no origin. It is absent in all Bat viruses and all Pangolin viruses. It is impossible to overstatement the significance of this feature. So the “location” of this feature cannot have been accidental. There are only two conclusions; either someone in a lab put it there or GOD himself stepped in to destroy evolution and genetics. To find this feature in this virus right from the start and not be able to determine the original host animals… you have a better chance of winning the lottery 50 times. Nature does not work like this. For clarification, this superman feature is missing in both SARS and MERS.
This feature superpowers the virus in terms of how it can spread in cells. The Flu, Common cold, Ebola, Zika, and Yellow Fever all have this. We both know how contagious and rampant the flu and common cold are. The existence of this within nCoV-2019 means this is a super virus.
Hopefully this has been helpful, I tried to keep it non-technical. ![]()
COVID-19, SARS and Bats Coronaviruses Genomes
Unexpected Exogeneous RNA Sequences
By Luc Montagnier, 2008 Nobel Prize in Physiology or Medicine for his discovery of the human immunodeficiency virus.
Jean-Claude Perez, PhD Maths § Computer Science Bordeaux University, RETIRED interdisciplinary researcher (IBM Emeritus, IBM European Research Center on Artificial Intelligence Montpellier), Bordeaux metropole, France
https://osf.io/d9e5g/
"We cannot apply the current and proven tools and methods of fighting against viruses, hitherto NATURAL, because we do not know today how a new virus whose part of the genome is SYNTHETIC in nature will evolve.
- This was already true for SARS, and even more so for COVID-19."
- This preprint was last updated 4.25.20. I would love your comments. TR
There are indeed a number of residues in this virus that present HIV homology. This is how I like to describe this virus in non-technical terms.
For our example, we will call the virus a car. By virus though I am very specifically talking about aspects of the Spike protein. Each car in this example represents a different protein in the human body that naturally serves different purposes. Through extended evolution (years or decades) a virus can gain new abilities and learn to deal with new cars.
SARS-CoV was like a thief. It could only target Honda cars. It then had to break the car window and manually attempt to hotwire the car. Not very efficient and this was only working some of the time.
MERS-CoV had the same problem with Hona cars but had the keys to Toyota cars. With Toyota cars, it could unlock the car, turn on the ignition, and drive away.
hCoV-2019 came with keys to 6+ car brands. This is beyond suspicious. This virus does not need to hotwire these cars. Sometimes the key does not work properly but let’s say it has many sets of Honda keys, so it can use many attempts to hijack that car. This is a super virus. One of the cars is called CD4. Without going into detail, let’s call this one of the primary T-cells for the immune system. HIV uses its GP160 protein receptor as a car key to enter CD4 and infect the host with HIV. While hCoV-2019 cannot give a host HIV nor does the virus contain traces of the HIV (virus), it has the “same” car key to access CD4. This is what Dr. Montagnier and his team were examining in their research. We believe a CD4 receptor hijack would weaken the immune system of the host and the host would potentially both test false-positive for HIV and present with “HIV-like symptoms” in the sense of immune deficiency.
This is very important research and only a few around the world are taking this seriously. Why? Because no scientist wants to admit the reality that this virus came with 6 car keys.
Everyone needs to avoid this virus as best they can. This is not a respiratory virus like SARS but a honey badger virus that can and will try to attack every organ in your body until you submit.
For a non-technical person, that analogy gives me a starting point to understand what you are talking about. Thank you.
My understanding of what you just said:
The "keys" in this analogy are receptor docking and access capabilities. The "cars" are different cell types.And this nCoV-19 is able to enter many different cell types.
nCov-19 is like a highly trained thief who was taught this trade by a team of master thieves called in to train him how to break into each different cell type. This concentration of expert thievery skills (into so many different cars) does not occur naturally.
Re: Post #67
”…The Malaysian Pangolin virus in question is actually pCoV-MP789 (MT121216) which was sampled from a wild Pangolin in southern Yunnan province, quite close to the Malaysian border. …”
Just want to clarify something. Minor but important to me at least. Not downplaying Dr Mayer’s post at all. The Malayan pangolins are also found in Myanmar which borders Yunnan. Malaysia is way way further down from Yunnan. There’s Thailand that borders Malaysia.
Dr. Mayer - thank you for introducing yourself in the forum, and for contributing your analysis to this conversation.
I’m not sure if this is outside the scope of your work, but given the research of this virus by you and your colleagues, do you have any comments on the subject of immunity to the virus, and also the expectation of potential mutations (including the question of immunity to new mutations, for those who were exposed to an earlier strain) ?
Thanks for the correction! That jogged my memory. I had previously stated that the Pangolins were found in Yunnan, next to Malaysia. Incorrect and I was thinking of a different case. For the Pangolin(s) in question, they were being smuggled into China and were seized as Guangdong customs. Sequencing analysis was completed in March and July of 2019 . All three smuggled Pangolins died from severe respiratory diseases.
Not to scare everyone but a vaccine for this virus is highly improbable. We could not develop a vaccine for SARS, even after 10 years of research. This virus is at least 10x worse. Not the information that people want to hear but our best chance from a scientific angle is to find ways to slow the virus down to reduce symptoms. Everyone wearing masks for an extended period would slowly starve the virus out.
RNA viruses tend to have many mutations and we have been tracking these in great detail. The problem has been with the lack of sequencing. PCR testing or RdRp sampling cannot determine if a patient has a key mutated version of the virus. Globally, we have genome data on roughly 0.6% of confirmed cases. There are 7 key “groups” of this virus and more will appear over time. I am not referring to mutations but something called clades. Virus-clades-mutations, that gets branched out

L, S, V, O are clades that have their mutations in the ORF1ab protein of the virus and these various changes can cause patients to present with different symptoms. Korea, Italy and UK, for example, were hit by the V clade. France initially had O and then L. In February we then saw a key mutation in the Spike protein which caused the virus to become more deadly. This is the G clade. GR and GH are essentially G+R or G+H. Aside from having the key (G) Spike mutation these genomes also have (different) mutations in the N protein. Of the genomes that we have on file:
GR: 16,451
G: 13,099
GH: 12,343
S: 4,017
V: 3,727
L: 3,445
(Other: 2,753)
*Keep in mind that many countries around the world are not reporting their cases and therefore we are not receiving any genome information. So the global cases and deaths are exceptionally skewed and incomplete.
G+ is currently responsible for 70% of sequenced genomes. Please keep in mind that these numbers only represent 0.6% of cases.
As with influenza if a host was infected with strain A, develops anti-bodies then the host would be less susceptible to strain A within X time period. The host would still be susceptible to strain B. This is because each strain has its own unique RNA signature and your immune system has a memory for each signature. A better example is that Border patrol would look at two brothers entering customs. They know to exclude brother 1 (antibodies) but they allow brother 2 to enter because his social security number is different. In reality, these two are not brothers but the same virus, posing as two different “people”. Hopefully that makes sense. We are finding that hosts are not currently developing long-lasting immune responses to this virus. This means there is even less likelihood to develop antibodies for A and be protected from B. The good news is that since G+ is the current dominant clade, developing any antibodies against these strains is much better compared to L, V, S, O.
You are getting the basis of this, but one important point of clarification
“nCov-19 is like a highly trained thief who was taught this trade by a team of master thieves called in to train him how to break into each different cell type.”
This situation is far worse as the virus came packaged having already mastered all of these aspects. To say this is highly unusual for a virus is a massive understatement. As you suggested, the virus needs to learn a skill and it becomes better over time. How then could this virus be a master from the start? This is one of the reasons why saying this virus naturally “evolved” does not work very well since it appears to have skipped many generations of evolution.